
Page 226 - The Vasculitides, Volume 1: General Considerations and Systemic Vasculitis
P. 226
202 Loic Guillevin
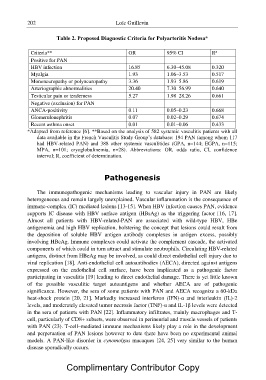
Table 2. Proposed Diagnostic Criteria for Polyarteritis Nodosa*
Criteria** OR 95% CI R2
Positive for PAN 16.85 6.30–45.08 0.320
HBV infection 1.93 1.06–3.53 0.517
Myalgia 3.36 1.93–5.86 0.619
Mononeuropathy or polyneuropathy 20.40 7.30–56.99 0.640
Arteriographic abnormalities
Testicular pain or tenderness 5.27 1.98–28.26 0.661
Negative (exclusion) for PAN 0.11 0.05–0.23 0.668
ANCA-positivity
Glomerulonephritis 0.07 0.02–0.29 0.674
Recent asthma onset
0.01 0.01–0.06 0.433
*Adapted from reference [6]. **Based on the analysis of 582 systemic vasculitis patients with all
data available in the French Vasculitis Study Group?s database: 194 PAN (among whom 117
had HBV-related PAN) and 388 other systemic vasculitides (GPA, n=144; EGPA, n=115;
MPA, n=101; cryoglobulinemia, n=28). Abbreviations: OR, odds ratio, CI, confidence
interval; R, coefficient of determination.
Pathogenesis
The immunopathogenic mechanisms leading to vascular injury in PAN are likely
heterogeneous and remain largely unexplained. Vascular inflammation is the consequence of
immune-complex (IC) mediated lesions [13-15]. When HBV infection causes PAN, evidence
supports IC disease with HBV surface antigen (HBsAg) as the triggering factor [16, 17].
Almost all patients with HBV-related-PAN are associated with wild-type HBV, HBe
antigenemia and high HBV replication, bolstering the concept that lesions could result from
the deposition of soluble HBV antigen–antibody complexes in antigen excess, possibly
involving HBeAg. Immune complexes could activate the complement cascade, the activated
components of which could in turn attract and stimulate neutrophils. Circulating HBV-related
antigens, distinct from HBeAg may be involved, as could direct endothelial cell injury due to
viral replication [18]. Anti-endothelial cell autoantibodies (AECA), directed against antigens
expressed on the endothelial cell surface, have been implicated as a pathogenic factor
participating in vasculitis [19] leading to direct endothelial damage. There is yet little known
of the possible vasculitic target autoantigens and whether AECA are of pathogenic
significance. However, the sera of some patients with PAN and AECA recognize a 60-kDa
heat-shock protein [20, 21]. Markedly increased interferon (IFN)-? and interleukin (IL)-2
levels, and moderately elevated tumor necrosis factor (TNF)-? and IL-1ß levels were detected
in the sera of patients with PAN [22]. Inflammatory infiltrates, mainly macrophages and T-
cell, particularly of CD8+ subsets, were observed in perineurial and muscle vessels of patients
with PAN (23). T-cell–mediated immune mechanisms likely play a role in the development
and perpetuation of PAN lesions however to date there have been no experimental animal
models. A PAN-like disorder in cynomolgus macaques [24, 25] very similar to the human
disease sporadically occurs.
Complimentary Contributor Copy