- Posted by admin
Neurologic hereditary metabolic disorders
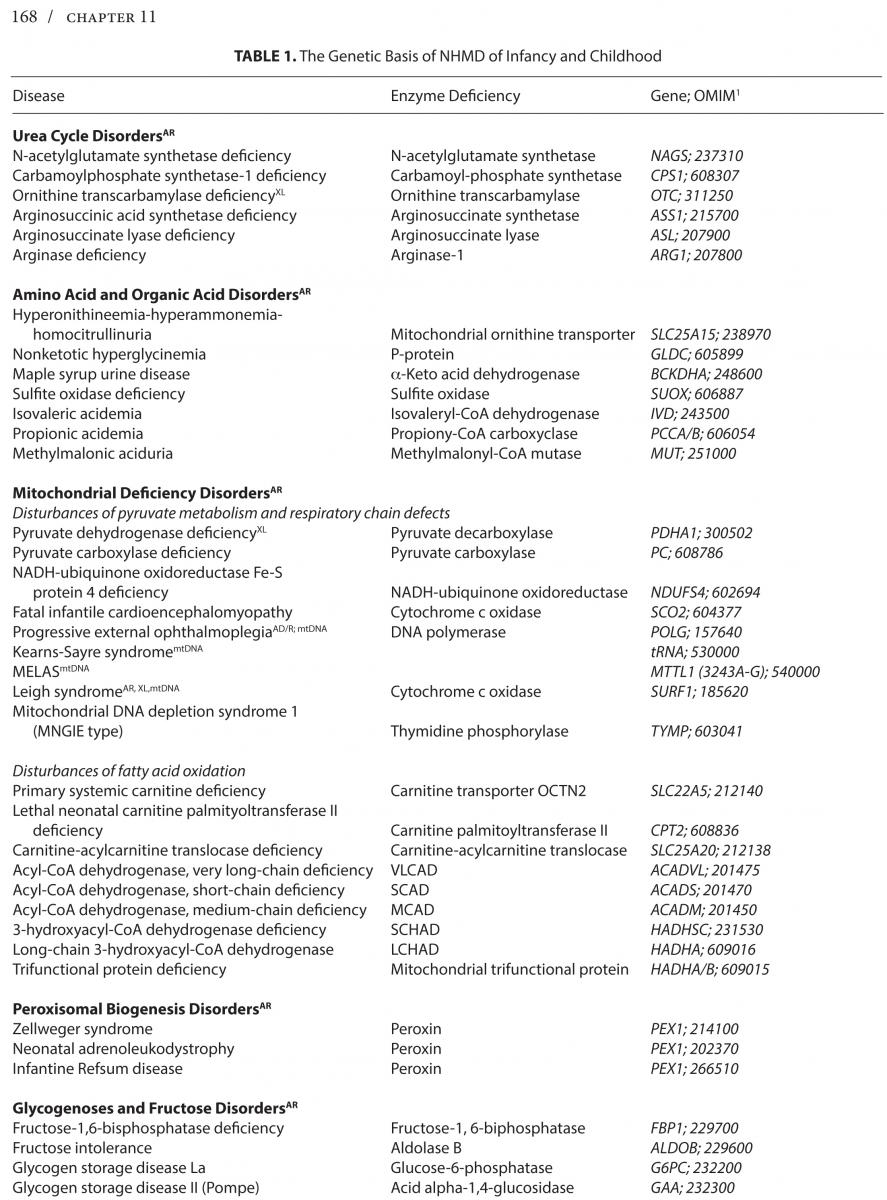
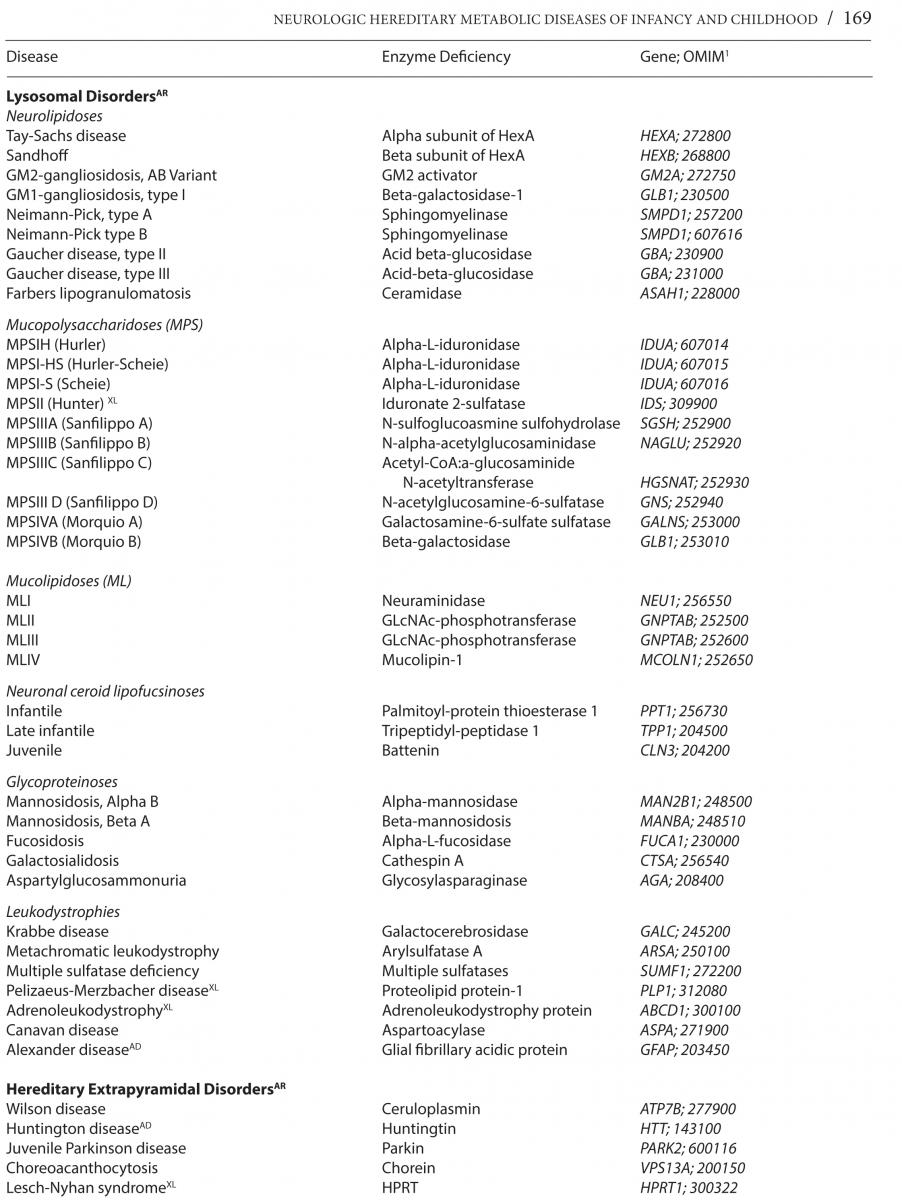
Neurologic hereditary metabolic disorders (NHMD), while difficult to recognize at birth, lead to unmistakable neurological symptoms and signs that become clinically evident in the neonatal period, through early and late infancy into childhood, each with a specific biochemical, molecular or cytogenetic defect, and unique genetic inheritance pattern. Even though each of the NHMD is exceedingly rare, the group as a whole is not, with an estimated prevalence of 1 in 5,000 children. *Table 1 lists the known NHMD along with the associated enzymatic deficiency and ascribed neurogenetic defect.
CLASSIFICATION
The vast number of NHMD can be categorized into one of three possible disturbances causing: 1) Storage of large molecule metabolism such as glycolipids and glycoproteins as occurs in lysosomal, leukodystrophy, and peroxisomal storage disorders affecting multiple visceral organs and the nervous system over time; 2) Altered small molecule metabolism as, for example, amino acids as in amino acid and organic acidurias, and urea cycle disorders that lead to early acute toxicity; 3) Glycolytic, fatty acid, and mitochondrial metabolic disturbances with deficient energy production resulting in slowly progressive and static motor disturbances and intermittent crises. Another method of classification is by age at onset from neonatal to early and late infancy or childhood. The genetic basis of the NHMD includes autosomal recessive, dominant, X-linked, and mitochondrial DNA inheritance.
NEUROLOGICAL ASSESSMENT
The neurological assessment of infants and young children can be challenging. Systemically addressed, it can provide helpful information in the diagnosis of a NHMD, and in identifying patients likely to benefit from early intervention. The available methods to assess and objectify motor function from birth to six years vary widely and differ in validity and reliability. The most accurate assessment is achieved through a combination of the neurological examination, achieved milestones, and assessment of the quality of motor behavior.
NHMD IN DIFFERENT STAGES OF CHILDHOOD
Clues to the presence of a NHMD in the neonatal period may occur in a low Apgar score at birth, and impaired alertness. There can be pupillary changes, impaired visual acuity, and oculomotor function leading to altered visual fixation and tracking, as well as, unusual postures and automatisms of the trunk and limbs, impairment of the Moro response, and abnormal supporting, placing, stepping reactions, palmar grasping, and tonic neck reflexes. Reduced sucking and swallowing mechanisms and seizures may occur, the latter presenting clinically with momentary apnea, tonic turning, posturing, eye and limb jerks, flexor and extensor spasms. Electroencephalography may show more severely disorganized background rhythms, multifocal and periodic bursts of epileptic activity, and intervening depressions that correspond to clinical flexor spasms. Associated findings suggestive of an NHMD include disturbance of the respiration, a peculiar odor of the urine, cardiomyopathy, organomegaly, polycystic kidneys; dysmorphic facial features, micro- and macrocephaly, skeletal changes, disturbed skin and hair, and ocular abnormalities such as cataracts, corneal opacities, and retinal degeneration. Hypoglycemia, metabolic acidosis, and ketosis are features suggestive of an aminoacidopathy, defect in biotin metabolism, and disturbances in carbohydrate metabolism. Markedly elevated ammonia levels and respiratory alkalosis occur in urea cycle disorders. Lactic acidosis in blood and cerebrospinal fluid are clues of an underlying mitochondrial disorder affecting pyruvate and respiratory chain reactions, while hypoglycemia and ketone bodies are seen in disorders of mitochondrial fatty acid oxidation. Elevated saturated very long-chain fatty acids suggest a lipidosis, that belong to one of two groups, either due to virtual absence of peroxisomal function with severe consequences, and another less severe phenotype with single enzyme deficiencies of b-oxidation. Suspicion of an early infantile NHMD in the first year of life arises in infants with neurological regression, delayed or arrested development, and lack of visual interest in their surroundings, as well as poor head control, inability to roll over, sit unsupported, or productively use the hands due to poor motor control. There may be a macular cherry red spot, macular degeneration, optic atrophy, retinal degeneration, nystagmus, and eye palsy. Other important neurological phenomena may include rigidity, reduced muscle tone, peripheral neuropathy, chorea, dystonia, and ataxia.
SUMMARY
An appreciation of these disorders is essential to a clear understanding of the natural history, prognosis, and therapeutic decisions, including genetic counseling for prenatal diagnosis and carrier detection, and in the selection of potentially effective treatments to preserve neurological function and forestall deterioration.
__________________
*Neurologic hereditary metabolic disorders of infancy and childhood. Chapter 11, Motor Disorders, 2015.